متلازمة أنجلمان (AS) هو اضطراب النمو العصبي تتميز صعوبات تعلم شديدة، وترنح، واضطراب الحجز مع EEG مميزة، ملامح الوجه تشوه خفية، وسعيد، والتصرف مؤنس. معظم الأطفال الموجودين مع تأخير في المعالم التنموية وتباطؤ النمو في الرأس خلال السنة الأولى من الحياة. في معظم الحالات الكلام لا تتطور. المرضى الذين يعانون من AS يكون النمط الظاهري السلوكية مميزة مع الحركات متشنج، والضحك المتكرر وغير مناسب في بعض الأحيان، حب من الماء، واضطراب النوم. ملامح الوجه خفية وتشمل نطاق واسع، يبتسم الفم والذقن البارز وعميقة تعيين العينين. وهو ناتج عن مجموعة متنوعة من التشوهات الجينية التي تنطوي على المنطقة كروموسوم 15q11-13، الذي يخضع ليطبع الجينوم. وتشمل هذه الحذف الأمهات، disomy أحد الأبوين الأب، وعيوب الطباعة، والطفرات نقطة أو الحذف صغيرة داخل الجين
UBE3A، التي تقع في هذه المنطقة. يظهر UBE3A يطبع الأنسجة محددة، يجري التعبير عنه حصرا من أليل الأمهات في الدماغ. تم العثور على الآليات الوراثية التي تم تحديدها حتى الآن في AS في 85-90٪ من أولئك الذين لديهم النمط الظاهري السريرية وجميع تتداخل مع UBE3A التعبير.
في عام 1965، هاري Angelman، وهو طبيب أطفال الإنجليزية، حسبما ذكرت والنتائج السريرية في ثلاثة أطفال مع ميزات مشابهة لصعوبات التعلم الشديدة، رنحية والحركات متشنج، وعدم القدرة على الكلام، وسهولة أثار الضحك. الثلاثة جميعهم نوبات الصرع مع ظهور EEG مميزة وملامح الوجه تشوه خفية.
1 هذا الشرط، في الأصل المعروف باسم متلازمة "دمية سعيدة"، ومن المعروف الآن بمصطلح أقل تحقير من متلازمة أنجلمان. على مدى 20 عاما وذلك بالنظر في اضطراب نادر، وعلى الرغم من وقوع الأسر التي لديها sibs المتضررة اقترح المسببات المرضية الوراثية، يمكن أن يكون في البداية تحديد أي سبب معروف. في عام 1987 Magenis
آخرون 2 حدد حذف من كروموسوم 15q11-13 في اثنين من المرضى الذين يعانون من متلازمة أنجلمان والأعمال اللاحقة أثبتت أن متلازمة أنجلمان يمكن أن تسببها مجموعة متنوعة من الآليات الوراثية التي تنطوي على هذه المنطقة مطبوع من الجينوم. كل من هذه الآليات تؤثر على التعبير الأمهات من الجينات
UBE3A التي تقع في موضع 15q11-13.
3 وفي السنوات الأخيرة أدى ترسيم أكثر وضوحا من النمط الظاهري السريرية لمتلازمة أنجلمان وتحسين الاختبارات التشخيصية لتحسين الاعتراف حالة وقوع Angelman ويقدر الآن متلازمة ما بين 1 في 10 000 و 1 في 40 000.
4- 7
دراسات من السمات المعرفية والسلوكية المحددة المرتبطة AS
8 تحسنت واضطراب الاستيلاء إدارة الحالة وتوفير نظرة ثاقبة في التوقعات على المدى الطويل للمرضى المتضررين. وقد بدأت الدراسات الجينية الجزيئية لتوضيح دور الجينات في المنطقة 15q11-13 في الفيزيولوجيا المرضية لمتلازمة أنجلمان
9 وألقت الضوء على ظاهرة أعم من يطبع الجينومية.
10 ومراجعة المظاهر السريرية، والتاريخ الطبيعي، والتيار إدارة الحالة وتلخص الآليات الوراثية المعقدة المطروحة.
المظاهر السريرية:
المرضى الأصلي أوردته Angelman
1 زيارتها عن العجز الشديد التعلم، ونوبات الصرع، وترنح، والكلام غائبا، وملامح الوجه تشوه مع ذقن بارزة وعميقة تعيين العينين والفم واسعة مع جاحظ اللسان، وصغر الرأس مع قفا شقة. كانت ناقصة الصباغ هم أيضا مع الشعر الأبيض والعيون الزرقاء (الشكل 1). على الرغم من أن العديد من المرضى الذين يعانون من AS يكون لهذه الخصائص،
11 هو عليه الآن واضحا أن الطيف السريري من متلازمة أنجلمان هو أوسع بكثير مما كان يعتقد أصلا. المصطبغة عادة العديد من المرضى وبعضها محيط الرأس الطبيعي. المضبوطات غير موجودة في كل حالة، وربما تكون بعض الكلام الحالي. بعض المرضى قد لا يكون سحنة تشوه مميزة ويكون الحد الأدنى ترنح. العديد من المرضى الذين يعانون من AS قادرين على الكلام، وعلى الرغم من أن الكلام هو دائما محدودة. الميزات السلوكية ينظر في AS ولعل ميزة السريرية أكثر اتساقا. فمن الضروري، بالتالي، أن يكون هناك مؤشر واسعة من الشك السريري في الطفل مع النمط الظاهري السلوكي للحالة كما هو موضح أدناه. ويرد موجز من السمات الرئيسية السريرية وتردداتها في الجدول 1.
الجدول 1 الخصائص السريرية الرئيسية للAS
 الشكل 1
الشكل 1 (A) AS المريض مع 15q11-13. (B) AS المريض مع disomy أحد الأبوين. (C) AS المريض مع عيب يطبع. (D) AS المريض مع الطفرة UBE3A.
الخصائص السلوكية للAS لافتة للنظر وهذه هي التي غالبا الأطباء الفوري للنظر في التشخيص. كانت موجودة في جميع المرضى بغض النظر عن نوع من الشذوذ الجيني. تبدأ نوبة من الضحك أثار بسهولة في غضون الأسابيع القليلة الأولى من الحياة وتقريبا جميع المرضى سعداء وابتسامة في كثير من الأحيان. وعادة ما أثارت الضحك، ولكن التحفيز في كثير من الأحيان الحد الأدنى والضحك يمكن أن تكون غير لائقة. فرط الحركة واضطراب النوم شائعة في مرحلة الطفولة، ويمكن أن تثير مشاكل الإدارية الرئيسية. يمكن تحسين اضطراب النوم عن طريق العلاج السلوكي مع التمسك نظام صارم وقت النوم واستخدام الميلاتونين، وهو فعال في حوالي 50٪ من المرضى.
12 الناس مع Angelman المياه متلازمة الحب ولها سحر لالسطوح العاكسة للمواد البلاستيكية والبالونات. انهم يتمتعون يجري في الشركة من الآخرين، ومشاهدة التلفزيون، وخاصة الفكاهة تهريجية. مع التقدم في مرحلة البلوغ يصبح السلوك زيادات أكثر هدوءا وتركيز العمر. التصرف مؤنس لا تزال قائمة وقد تحدث نوبة من الضحك. وقد أظهرت بعض الكبار الميل إلى السلوك العدواني، وخاصة إذا بالاحباط بسبب صعوبة الاتصالات.
صعوبات في الاتصالات هي سمة بارزة من سمات متلازمة أنجلمان.
13 كلام لا تتطور والأكثر AS المرضى الذين لديهم مفردات سوى اثنين أو ثلاث كلمات.
14 معظم المرضى سوف يكون قادرا على فهم أوامر بسيطة في إطار روتين حياتهم اليومية. وهناك أقلية يمكن الاتصال باستخدام لغة الإشارة الرسمية مثل ماكتون أو نظام الاتصالات تبادل الصور (PECS). آخرون استخدام الإيماءات للتواصل. وكانت بعض المرضى قادرين على استخدام أجهزة الاتصالات المدمجة (على سبيل المثال، شركة دينافوكس) إلى تأثير جيد واكتساب مهارات الاتصال غالبا ما يكون أسهل مع تقدم المرضى من كبار السن ويحسن القدرة على التركيز. صعوبات في التواصل يمكن أن يؤدي إلى الإحباط. الكلام موجود في عدد قليل من المرضى (انظر النمط الظاهري / النمط الجيني الارتباط)، ولكن حتى هذه سوف يجدون صعوبة في فهم الأوامر المعقدة وعدم وجود القدرة على التفكير المجرد.
تختلف مهارات المساعدة الذاتية في متلازمة أنجلمان. معظم المرضى تعلم المشي والتواصل يحب ويكره. ويمكن أن تجعل الخيارات، على سبيل المثال، فيما يتعلق الطعام الذي يأكلونه والملابس التي يرتدينها. هناك حاجة لمساعدة مع خلع الملابس والاستحمام، وعلى الرغم من تعريتها أسهل. التدريب على استعمال المرحاض هو ممكن، وحوالي ثلث المرضى سوف تكون جافة بعد يوم، وأقل ليلا. يمكن للناس مع AS تتغذى عادة أنفسهم باستخدام أواني الأساسية. كلها تتطلب الإشراف وأنها لا تملك الإحساس بالخطر. على الرغم من المرضى غير قادرين على العيش بصورة مستقلة، وكثير ديك نوعية جيدة من الحياة مع وجود شبه مستقلة مع جولة إشراف على مدار الساعة.
كل AS المرضى لديهم تخلف عقلي شديد والمعالم السيارات تأخر. يجلسون غير معتمد في حوالي 12 شهرا، والزحف (نمط الكوماندوز) أو خلط السفلي في 18-24 شهرا، والسير في متوسط العمر 4 سنوات (المدى 18 شهرا إلى 7 سنوات).
5 ومشية بطيئة، وقاسية الأرجل، و رنحي وتربى في أحضان وعقدت استعرضوا في المعصمين والمرفقين. ترفرف اليد هو شائع عند المشي، وإذا متحمس. العضلات غير طبيعي مع نقص التوتر جذعي وفرط في الأطراف وردود الفعل هي سريعة. يحدث الجنف الصدري في ما يقرب من 10٪ من الأطفال، ولكن مشكلة رئيسية في معظم المرضى البالغين. قد تحتاج إلى علاج العظام عن طريق العلاج الطبيعي أو تستعد وعلى الرغم من أن هذه قد تكون كافية لحل الجنف في بعض المرضى، في المرضى الآخرين التشوه هو تقدمي ومطلوب لعملية جراحية. كثير من المرضى الذين يعانون من AS يكون الحركات متشنج وهزة منفصلة من الأصابع بسبب رمع عضلي القشرية. وتفاقمت هذه الظاهرة بسبب الإجهاد وأكثر وضوحا لدى البالغين، وبعضهم وضع زيادة الزلزال.
تحدث نوبات الصرع في 80٪ من المرضى. العمر عند بداية يتراوح بين سنة وخمس سنوات. العرض الأولي لمرض الصرع هو مع التشنجات الحموية في سن الطفولة.
15، 16 في مرحلة الطفولة مجموعة متنوعة من نوبات يمكن ملاحظتها، بدءا من النوبات التوترية الارتجاجية، الغياب شاذة ومعقدة الجزئي، الرمع العضلي، واهن، والمضبوطات منشط لحالة صرعية. قد يحدث أيضا حالة الغياب ووضع رمعية. وأشارت تقارير سابقة تردد تقليل من نوبات الصرع مع التقدم في السن،
5، 17، 18 ولكن كذلك متابعة أظهرت أنه على الرغم من أن تكون هناك فترة هادئة نسبيا خلال مرحلة الطفولة المتأخرة والمراهقة، وعدد كبير من البالغين لديهم نوبات الصرع، الغياب شاذة ولا سيما أو المضبوطات الرمع العضلي.
19 والصرع من متلازمة أنجلمان هو من الصعب السيطرة عليها بالأدوية المضادة للصرع، وخاصة في مرحلة الطفولة. الأدوية الأكثر فعالية هي فالبروات الصوديوم كعلاج وحيد أو بالاشتراك مع كلونازيبام أو غيرها من البنزوديازيبينات. كاربامازيبين له أحيانا أثر سلبي.
19، 20 في البالغين، الفينوباربيتون فعال أيضا. تجربة مع العقاقير المضادة للصرع أحدث محدودة للغاية.
هناك أنماط EEG محددة في AS المرضى التي تظهر في عزلة أو في مجموعات مختلفة. وهي تشبه في المرضى الذين يعانون من نوبات ودون حد سواء.
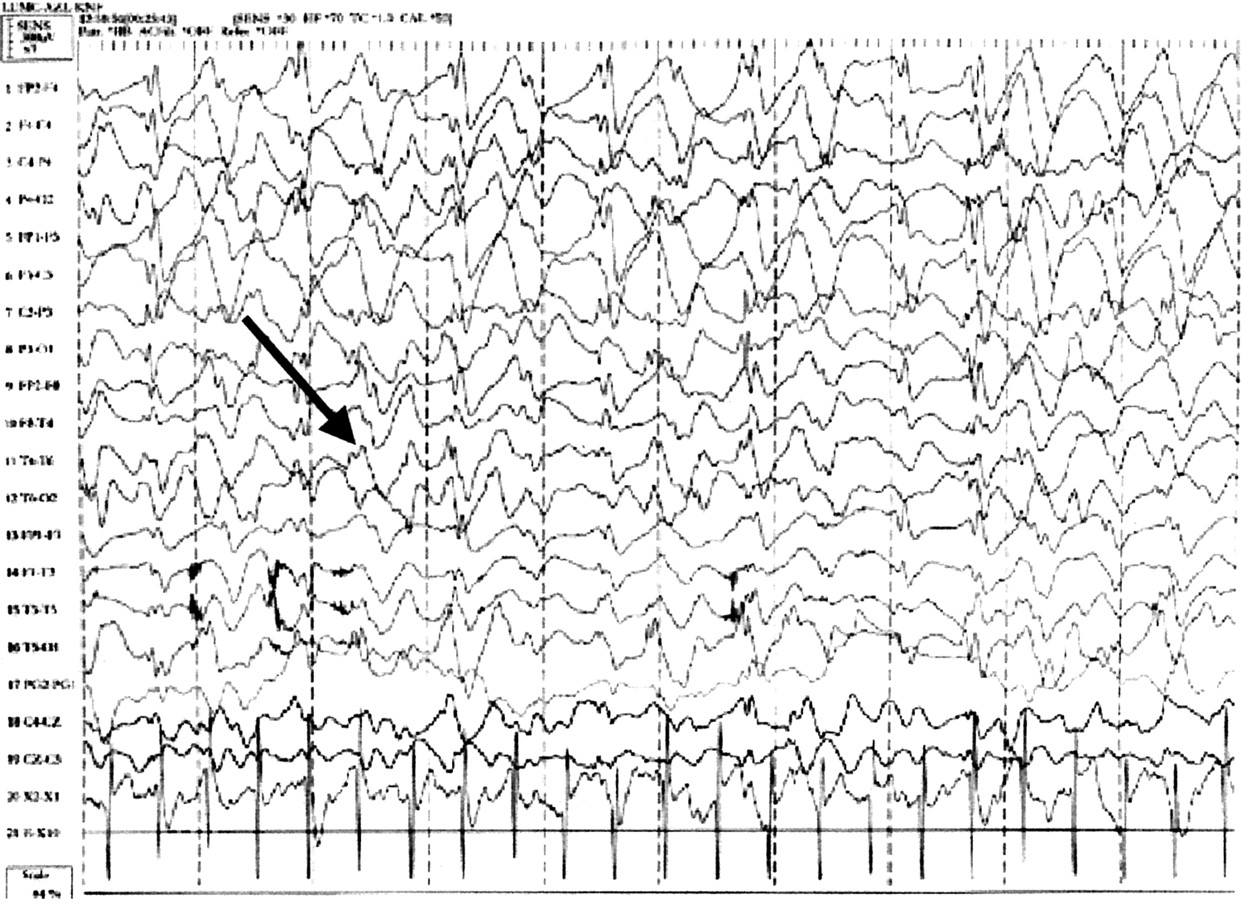
20، 21 في مرحلة الطفولة أنماط مميزة الثلاثة هي (1) استمرار الإيقاعية 4-6 / ق النشاط تصل إلى أكثر من 200 μV، لا ترتبط مع النعاس، (2) أشواط طويلة من الإيقاعي (ثلاثي الأطوار) 2-3 / ق النشاط مع اتساع 200-500 μV، القصوى فوق المناطق الأمامية ويخلط عادة مع المسامير أو موجات حادة، و (3) المسامير مختلطة مع 3-4 / ق المكونات، عادة أكثر من 200 μV أساسا الخلف ويسرتها أو ينظر إليها فقط على إغلاق العين. في المرضى الذين يعانون أكثر من 10 سنة من العمر إيقاع خلفية أبطأ من المعتاد. قد يكون هناك طفرات التنسيق والنشاط دلتا ثلاثي الأطوار متقطع أو مستمر (الشكل 2)، القصوى فوق المناطق الأمامية.
15 ليست هناك علاقة بين نتائج EEG ونوبة من الضحك ينظر في AS. الميزات EEG هي محددة لAS، وهي ميزة التشخيص الهامة. للأسف، قد لا يكون دائما ينظر إلى هذه العناصر الثلاثة في نفس تسجيل EEG ويمكن أن تحدث في أوقات مختلفة في نفس المريض.
15، 18، 22 وبالتالي يستحق تكرار EEG إذا كان يشتبه في تشخيص AS بقوة ولكن المظاهر التقليدية لا ينظر في البداية.
 الرقم 2
الرقم 2 EEG Angelman مميزة. arrowed ثلاثي الأطوار دلتا موجة النشاط.
النمط الظاهري من متلازمة أنجلمان هو واحد المتطور الذي يتغير مع التقدم في مرحلة البلوغ (الشكل 3). يحدث البلوغ في الوقت الاصلي وهناك الخصائص الجنسية الثانوية العادية. هي أكثر وضوحا خصائص الوجه عند البالغين المصابين فقم الفك السفلي ملحوظ، وأشار الذقن، شدق، وشفة السفلية البارزة.
17، 19، 23، 24 عيون يبدو تعيين أكثر عمقا وعند بعض المرضى القرنية المخروطية (بسبب فرك العين) لوحظ. التنقل النقصان بسبب فرط أطرافهم، وتطوير الجنف الصدري، وعدم الرغبة في المشي.
19 يعاني بعض المرضى من التقلصات وتلك مع انخفاض ترنح التنقل هو أقل وضوحا. كثير من المرضى يصابون قد يحدث ارتداد يعانون من السمنة المفرطة والمريء ويمكن أن تكون شديدة. وعلى الرغم من هذه المشاكل والحفاظ على نوعية جيدة من الحياة حتى سن البلوغ وليس تخفيض غالبية المرضى عمر إلى حد كبير.
 الشكل (3)
الشكل (3) ملامح الوجه من متلازمة أنجلمان في البالغين الصغار.
هناك أربع آليات وراثية الرئيسية التي تعرف الآن أن تسبب متلازمة أنجلمان وAS تم تقسيم المرضى إلى فئات من الأول إلى الرابع على أساس هذه الآليات.
3 مجموعة أخرى من المرضى، والطبقة V المعينة، ويكون المظاهر السريرية لAS لكن لا راثي خلوي إثباته أو الشذوذ الجزيئي للكروموسوم 15q11-13. ويرد موجز لمختلف الآليات الوراثية وتردداتها في الجدول 2 ويرد تمثيل شكلي للمنطقة 15q11-13 في التين 4.
الجدول 2 الآليات الوراثية مما يؤدي إلى متلازمة أنجلمان
 الرقم 4
الرقم 4 و/ المنطقة برادر ويلي Angelman على الصبغي 15q11-13 الذي يمتد 4 ميجا بايت. وتتمثل نقاط التوقف المشتركة بخطوط متقطعة. وصفت الجينات مطبوع أبويا في الجينات الزرقاء ومطبوع أمهات باللون الوردي. تلك المصورة في الأبيض لا يعرف أن يكون مطبوع. مركز يطبع (IC) هو الثنائي. الجزء القسيم المركزي هو المسؤول عن الأب لمفتاح بصمة الأم والحذف في هذا الجزء السبب AS. لمزيد من التفاصيل انظر النص.
الآلية الوراثية شيوعا مما أدى إلى متلازمة أنجلمان، وتحدث في حوالي 70-75٪ من المرضى، هو الحذف الخلالي من كروموسوم 15q11-13. غالبية الحذف ذات حجم مماثل، ما يقرب من 4 ميغابايت، ومع نقاط التوقف المشتركة. ويعتقد أنها تحدث بسبب عبور غير المتكافئ بين أكثر من يكرر نسخة منخفضة (duplicons) داخل المنطقة 15q11-13.
25 وduplicons تحتوي على نسخة من هذا الجين
HERC2، جين الحفظ جدا والتي عند تحور في الفئران، ويؤثر على التنمية والخصوبة .
26 وتحدث معظم الحذف دي نوفو وهي من أصل الأمهات، وعلى النقيض من 15q11-13 الحذف لوحظت في متلازمة برادر ويلي والتي هي من أصل أبوي.
27 ويمكن الكشف عن الحذف المشتركة من خلال تحليل FISH وعن طريق تحليل الحامض من SNRPN (صغير ببتيد بروتين نووي ريبوزي النووي N) المروج الذي يقع داخل جزيرة الدليل السياسي الشامل في 15q11-13. في وجود الحذف الأمهات فقط الأب، ونمط unmethylated يكون كشفها.
28 نادرا، المرضى الذين يعانون AS سيكون له حذف مختلفة الحجم، وغالبا بالتعاون مع النبات أو كروموسوم غير متوازن إعادة ترتيب.
29، 30 ومن المهم أن نميز هذا مجموعة من المرضى كما يجب على الأم أن تحمل نفس الصبغي إعادة ترتيب، وبالتالي تكون معرضة للخطر من وجود المزيد من الأطفال مع AS. تم الإبلاغ عن العديد من المرضى الذين كان الجرد الحزب الاتحادي الديمقراطي الزائدين (15) الحالي بالإضافة إلى الحذف من جديد من 15q11-13.
31 وقد افترض أن وجود مثل هذه علامة يهيئ ل15q11-13 الحذف. ولذلك ينبغي إجراء تحليل وراثي خلوي وتحليل FISH لاستبعاد النبات خفي على الناس من مع AS حيث طلب الأسر المعنية الاستشارة الوراثية.
الدرجة الثانية من AS المرضى، والطبقة الثانية، disomy أحد الأبوين (UPD) لكروموسوم 15، وبالتالي تفشل في وراثة نسخة الأمهات من
UBE3A. وUPD عادة ما يكون لكروموسوم كامل وروبنسون
وآخرون 32 درسوا في التفاصيل والآليات التي تنشأ فيها، وخلصت إلى أنه في معظم الحالات الخطأ هو تال للزيجوت، على الرغم من أن ثبت بعض الحالات أن تنشأ من خلال الانتصافي عدم انفصال. الأعمار الأم والأب من المرضى الذين يعانون من UPD هي أعلى بكثير من عامة السكان.
33 ومن المرجح أن تكون مسؤولة عن أعلى معدل لعدم انفصال في المرضى حيث نشأت UPD بسبب خطأ الانتصافي، ولكن الآلية التي أثارت الأب عمر يساهم في ازدواجية تال للزيجوت من كروموسوم 15 هو واضح. في بعض الحالات UPD (15) ينشأ في رابطة مع النبات روبرتسوني أو النبات المتبادل التي تنطوي على كروموسوم 15.
34، 35 وقوع disomy أحد الأبوين متقطع وحسابات فقط 2-3٪ من حالات AS.
الطبقة الثالثة المرضى هم دون حذف أو محدث، ولكن مع الشاذ كروموسوم 15 مثيلة، مما يدل على وجود خلل في يطبع. حساب عيوب يطبع لمدة 3-5٪ من المرضى الذين يعانون من AS. هذه هي العملية التي جينية وضع علامات على الكروموسومات يحدث في سلالة الجرثومية، بحيث أنها تحتفظ ذاكرة أصلهم الوالدين.
10 Buiting
وآخرون 36 حدد مركز يطبع داخل كروموسوم 15q11-13 الذي ينظم هيكل لونين، الحامض النووي، و التعبير الجيني من خلال عناصر التمثيل رابطة الدول المستقلة. هذا المركز يطبع (IC) لديها بنية ثنائية. يظهر جزء واحد أن يشارك في التحول بصمة الأب إلى بصمة الأمهات أثناء عملية تكوين الأمشاج، في حين أن الجزء الآخر هو المسؤول عن الأب إلى التبديل الأمهات. حوالي 50٪ من المرضى يعانون من عيوب يطبع لديها طفرة تعريفية داخل مركز يطبع، ولكن في بقية المرضى لا يمكن تحديد الطفرات. ومن المعتقد التي هي سبب في هذه المجموعة الأخيرة يطبع العيوب الأحداث ما قبل أو تال للزيجوت عفوية.
37 تقرير صدر مؤخرا عن كوكس
وآخرون 38 واقترحت أن حقن الحيوانات المنوية داخل الهيولى قد تكون آلية التي تتعارض مع إنشاء بصمة الأمهات في بويضة وبالتالي قد يؤهب لمتلازمة أنجلمان. من بين مجموعة من المرضى حيث تم الكشف عن الحذف مركز يطبع، وهناك العديد من الحالات الأسرية. حيث لم يتم تحديد الطفرات، كما هي عادة حدوث متفرقة. استثناء هي عائلة حيث كان موضوعات المتضررة والأمهات تتأثر انعكاس كروموسوم تحت المجهري في مركز يطبع.
39 للآباء الأطفال الذين يعانون من عيوب يطبع الذين يرغبون في مواصلة اختبارات ما قبل الولادة أثناء فترة الحمل في المستقبل، جلين
وآخرون 40 أظهر هذا التحليل مثيلة في
SNRPN موضع الحمض النووي المستخرج من زغابة المشيمي أو amniocytes سيعطي نتيجة موثوقة. كما تم افترض أن بعض المرضى الذين يعانون من المظاهر السريرية شاذة قد تكون فسيفساء لعيب مركز يطبع.
41 هذه المجموعة من المرضى غالبا ما تكون موجودة مع نقص التوتر والسمنة، وفي بعض الحالات من متلازمة برادر ويلي ومتلازمة لا Angelman التي يشتبه في البداية .
المرضى الذين يعانون من الدرجة الرابعة هم أولئك الذين ثبت لديهم طفرات في ترميز الجينات البروتين بتحول يغاز،
UBE3A. 42، 43 ويمكن تحديد الطفرات في 20٪ من المرضى متفرقة مع الحامض العادي وحوالي 75٪ من المرضى العائلي.
44، 45 هو مطبوع الجين
UBE3A في الدماغ ويشفر يغاز البروتين بتحول والتي يعتقد أنها تلعب دورا في ubiquitination من البروتينات في الدماغ، وهي العملية التي يصادف تلك البروتينات الموجهة للتدهور.
46 ويعبر عنه في الغالب داخل الحصين والخلايا العصبية لل يظهر المخيخ ويطبع محددة الأنسجة، يجري التعبير عنه فقط من أليل الأمهات في الدماغ ولكن مع التعبير biallelic في مكان آخر.
47 الفئران من نقص في Ube3a الأمهات لديهم ضعف المهارات الحركية والتعلم المكاني وعرض قراءات EEG غير طبيعية من منطقة قرن آمون.
48 وهناك عدة ركائز لUBE3A، ولكن تلك التي تلعب دورا حاسما في الفيزيولوجيا المرضية لAS لا يزال يتعين تحديدها.
9 يتكون الجين
UBE3A 16 الإكسونات مع الترميز المنطقة من الإكسونات 8-16. تم العثور على الطفرات نقطة في جميع أنحاء الترميز المنطقة بأسرها مع مجموعات في الإكسونات 9 و 16، وهذه الأخيرة التي تحتوي على نطاق وكان الحاخام هيشت الحفظ جدا. الشق الحفاز بين اثنين من فصوص المجال وكان الحاخام هيشت هو الموقع العديد من الطفرات عنها.
49 فحص المنطقة الترميز بواسطة SSCP تليها التسلسل المباشر هو وسيلة موثوقة نسبيا للكشف عن الطفرات. وقد تم تحديد انزياح الإطار، هراء، ولصق موقع الطفرات. كما تم الإبلاغ عن بعض الطفرات مغلطة. على الرغم من أنه من الصعب أن تكون على يقين فيما يتعلق بالحالة المرضية لهذه التغييرات الأخيرة، يجب على المرء أن يكون على درجة عالية من الشك حيث وقعت هذه من جديد أو تم الموروثة من الأم. كما تم تحديد العديد من الأشكال المشتركة داخل الجين
UBE3A. حيث تم تحديد الطفرات
UBE3A، يمكن تحليل الحمض النووي الأبوية لتحديد إذا كانت الأم تحمل نفس الطفرة. ويبدو أن معظم الطفرات تحدث دي نوفو وتشير تجربتنا أنه فقط حوالي 20٪ من الأمهات سيحمل نفس الطفرة. هناك، ومع ذلك، العديد من العائلات التي ذكرت حيث كان أمهات أكثر من الطفل المصاب على الرغم من زيارتها تحليل
UBE3A سلبي، ومن المرجح أن يكون الفسيفساء الغدد التناسلية هؤلاء الأمهات. لهذا السبب، ينبغي أن تقدم جميع أمهات الأطفال الذين يعانون من الطفرات
UBE3A اختبارات ما قبل الولادة في حالات الحمل المستقبلية. باستثناء عدد قليل من الطفرات التي تم العثور عليها في المريض أكثر من واحد، أكثر فريدة من نوعها. وأخيرا، وإن كان نادرا، برجر
وآخرون 50 أفاد المريض مع حذف 570 سنة مضت من
UBE3A الذي كان العائلية وتم الكشف من خلال فقدان أليلية في الصغرية مواضع. وهذا النوع من الشذوذ لم يكن للكشف على
UBE3A التسلسل.
لا تزال هناك بعض المرضى الذين يعانون من النمط الظاهري السريرية للAS حيث لم يتم التعرف على كروموسوم 15 شذوذ ويتم تعيين هؤلاء المرضى فئة V. على الرغم من أن بعض أزعم أن هؤلاء المرضى يجب أن يكون التشخيص بديلة، تجربتنا الخاصة، وذلك من جماعات أخرى لها خبرة سريرية كبيرة من AS، تشير إلى أن المرضى فئة V موجودة.
8 هذه المجموعة هو، ومع ذلك، من المحتمل أن تكون غير متجانسة وتشمل المرضى الذين يعانون من اضطرابات أخرى.
51- 58 بعض التشخيصات التي يجب استبعادها في هذه المجموعة هي متلازمة ريت،
59، 60 متلازمة التخلف العقلي-الثلاسيميا في α،
61 والحالة التي وصفها موات
وآخرون، والذي يعرف الآن أن تكون نتيجة الحذف كبيرة الحجم أو الطفرات نقطة في الجينات على الكروموسوم
ZFHX1B 2.
62، 63 تشخيص AS يمكن أيضا النظر في الأطفال الذين يعانون من أعراض مجمعات أوسع، مثل الشلل الدماغي، متلازمة لينوكس غاستو، أو اضطرابات الميتوكوندريا.
15 ، 22، 64 ويليامز
وآخرون 65 تلخيص العديد من الظروف التي تحاكي متلازمة أنجلمان ويعطى قائمة شاملة في الجدول 3.
الجدول 3 AS: التشخيص التفريقي
وتشمل تفسيرات أخرى لوجود هذه المجموعة إمكانية تحور
UBE3A داخل منطقة غير الترميز أو طفرة داخل جين آخر في مسار بتحول والذي يؤثر
UBE3A التعبير. ولم يتم تحديد أي طفرات حتى الآن داخل المنطقة 3 'الرئيسية
للUBE3A وليس هناك أي تقارير توثق حتى الآن التعبير عن
UBE3A داخل الدماغ للمرضى الدرجة V. وهناك نسخة من العقاقير
UBE3A 66 ولكن يتم التعبير عن هذا أبويا وغير معروفة الطفرات في هذا الجين للمشاركة في التسبب في AS. ومن بين المرشحين المحتملين، ومع ذلك، هو جين
ATP10C 67 والذي يقع في حدود 200 كيلو بايت من
UBE3A ولقد ثبت أن مطبوع للأمهات في الدماغ. حتى الآن، لم يتم تحديد أي طفرات داخل
ATP10C في البشر.
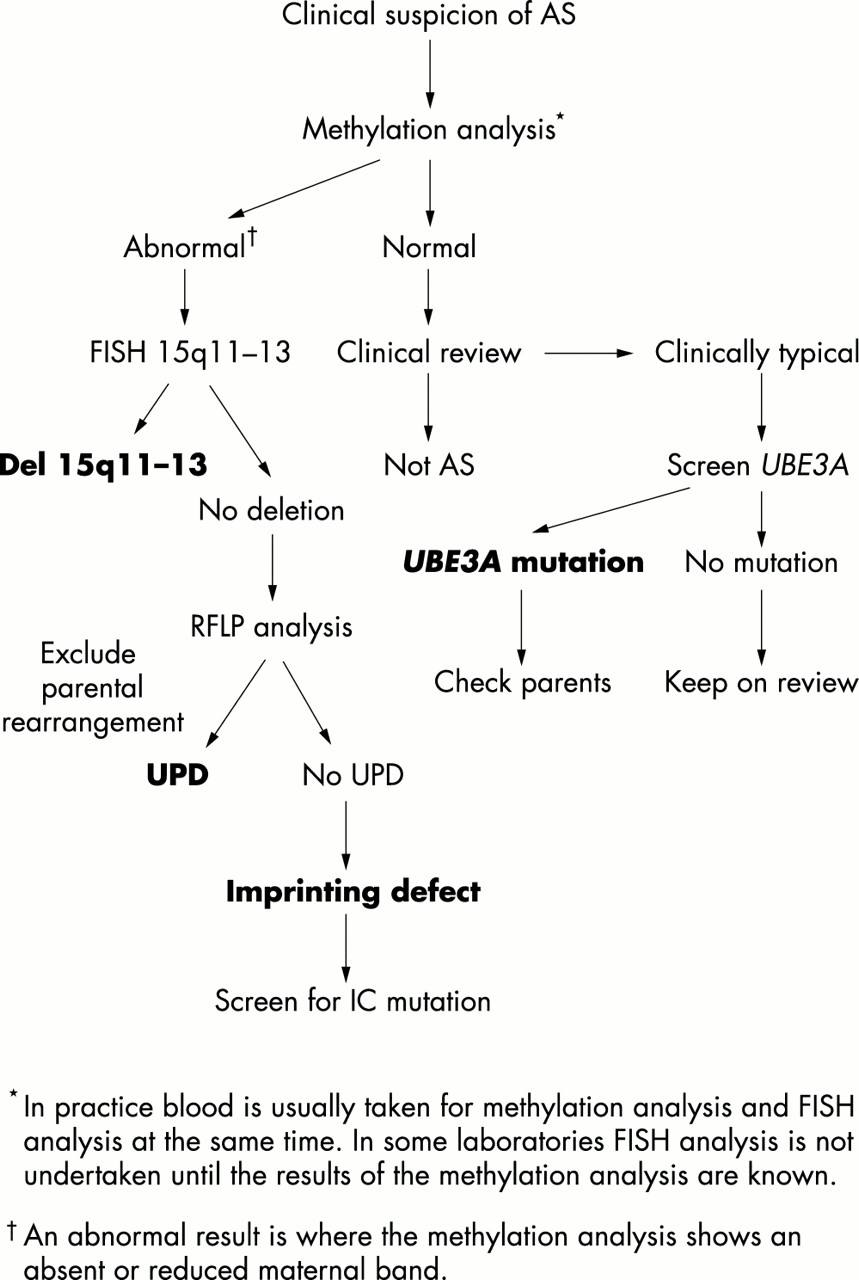
معقد الاستشارة الوراثية في الأسر AS بالنظر إلى تشكيلة واسعة من الآليات الوراثية التي يمكن أن تعطي كل ارتفاع لهذا الشرط. فمن المستحسن أن التحقيق التشخيص يجب أن تبدأ مع 15q11-13 تحليل الحامض، وهي غير مكلفة نسبيا، وسوف يكون غير طبيعي في أغلب الحالات من AS. يمكن للمرء ثم الانتقال إلى اختبارات أخرى لتوضيح آلية جينية محددة المعنيين أو للكشف عن الطفرات
UBE3A في المرضى الذين يعانون من ميزات موحية السريرية، EEG، والحامض الطبيعي. وأظهرت خوارزمية للاختبار الجيني في الشكل 5. مخاطر تكرار في AS سيعتمد على آلية الوراثية المعنية وحول ما إذا كان يتم عرض على الأم أن تحمل الشذوذ الجيني لل15q11-13. للمرضى الذين يعانون من جديد الحذف من 15q11-13 وdisomy أحد الأبوين، ومخاطر تكرار منخفضة، وإن كان هناك واحد الإصابة المبلغ عنها من تكرار نظرا لاحتمال الاصباغ الغدد التناسلية.
68، 69 أين هي المعنية الطفرات مركز يطبع والطفرات
UBE3A، فإن الوضع تعقيدا من مستويات عالية من الاصباغ الغدد التناسلية التي يبدو أنها موجودة في أمهات.
70، 71 وفي هذه الحالات، حتى إذا كانت الأم اختبارات السلبية لطفرة المحددة في طفل لا يزال هناك خطر ملموس من تكرار بسبب الاصباغ الغدد التناسلية وينبغي أن تقدم اختبارات ما قبل الولادة. إذا كانت الأم تحمل نفس الطفرة، من خطر تكرار هو 50٪. تم الإبلاغ عن تكرار ضمن عائلات المرضى فئة V والاستشارة الوراثية يحتاج لتعكس هذا. ومن المرجح أن تشمل بعض المرضى الذين يعانون من اضطرابات المتنحية X مرتبط أو مقهورة هذه المجموعة. في بعض العائلات وعرضت التشخيص قبل الولادة عن طريق تحديد المفردات الجنين أثناء الحمل في المستقبل لتحديد ما إذا كان الجنين قد ورث نفس الصبغي الأمهات 15 الحلقه الطفل المصاب. ومع ذلك، وهذا لا يمكن إلا أن عرضت في الأسر حيث التشخيص السريري للAS آمن جدا وينبغي بذل الآباء يدرك تماما من القيود المفروضة على هذا النوع من التجارب.
69، 72
 الرقم 5
الرقم 5 خوارزمية بسيطة لاختبار وراثي في متلازمة أنجلمان.
وقد ساعد ترسيم آليات وراثية مختلفة مما أدى إلى AS لشرح بعض الاختلافات المظهرية بين مجموعات المرضى (الشكل 1).
8، 41، 72- 76 فقد تبين، على سبيل المثال، أن المرضى الذين يعانون من كروموسوم 15 الحذف هم في عموما الأكثر تأثرا شديدا. لديهم نسبة أعلى من المضبوطات، وصغر الرأس، ونقص التصبغ، وزيادة تأخير في المراحل الحركية، والكلام غائبا. ويعتقد الميزات أشد أن يكون نتيجة لhaploinsufficiency لعدد من الجينات داخل المنطقة 15q11-13. ميناسيان
وآخرون 20 واقترح أن الصرع الحاد الذي تشهده AS المرضى الذين يعانون من الحذف هو نتيجة لعدم وجود نسخة واحدة من جينات مستقبلات
GABA. موضع
لP الجين الذي تورط في النوع الثاني من المهق عيني جلدي يكمن أيضا في هذه المنطقة.
77 وقد أظهرت الدراسات الحديثة أن AS المرضى الحذف مع نقص التصبغ يكون في كثير من الأحيان طفرة من
P الجينات على الكروموسومات نديد المتبقية
78، 79 وبعض المرضى لديهم نتائج العين مميزة من المهق مع misrouting من ألياف العصب البصري في chiasm البصرية.
80 المرضى الذين يعانون من disomy أحد الأبوين من كروموسوم 15 ديهم تدني نسبة المضبوطات، وصغر الرأس، ونقص التصبغ. العديد من هؤلاء المرضى قادرين أن أقول بضع كلمات والمعلمات نموها هي أيضا أكبر من مجموعة الحذف، ويجري في النصف العلوي من المعدل الطبيعي في كثير من الأحيان. ميزات تشوه أقل وضوحا في هذه المجموعة، على الرغم من أن الخصائص السلوكية هي نموذجية تماما. AS الموضوعات مع يطبع العيوب هي اقل عرضة لصغر الرأس، نقص التصبغ، أو المضبوطات، ومرة أخرى، هم أكثر قدرة من مجموعة الحذف مع أقل تأخير في المراحل الحركية ومهارات التواصل أفضل. نمو أفضل مما كانت عليه في المرضى الحذف والسمنة شائعة نسبيا ضمن هذه المجموعة. قدرات المرضى الذين يعانون من الطفرات
UBE3A تقع في مكان ما بين تلك المجموعة الحذف ومجموعة UPD. لديهم في كثير من الأحيان المضبوطات وصغر الرأس ولكن لم يتم ناقصة الصباغ ويكون أفضل المهارات الحركية والاتصالات من المجموعة الحذف. Lossie
آخرون 8 أشار إلى أن هذه المجموعة لديها عالية التردد لا سيما من السمنة كما يتقدمون في السن.
وأخيرا، أكثر اعتدالا AS تم الإبلاغ عن الظواهر في المرضى الذين يعانون من عيوب يطبع ناقصة أو الاصباغ. Gillessen-Kaesbach
وآخرون 41 وصف النمط الظاهري السريرية في سبعة AS المرضى الذين قدموا في البداية مع ملامح متلازمة برادر ويلي ولكن لم يظهر لاحقا أن يكون لها نمط مثيلة كروموسوم 15 وذلك تمشيا مع AS. هؤلاء المرضى قدم مع نقص التوتر، والسمنة، ودرجة أخف من التخلف العقلي. وأظهر تحليل الحامض في بعض من هؤلاء المرضى نمط شاذة مع وجود فرقة الأمهات باهتة. تكين
وآخرون 81 وأفادت أيضا المريض بمظاهر سريرية أكثر اعتدالا من AS الذين ثبت لديها الاصباغ لحذف 15q11-13، الذي كان يمكن كشفها في مضان التهجين في الموقع ولكن أين كان تحليل الحامض ثبت العادية.
إدارة AS تدور حول العلاجات المناسبة لمشاكل جسدية وعصبية واجه في هذه الحالة وتوفير لتلبية الاحتياجات التعليمية الخاصة، نظرا لغاية محددة الملامح المعرفية والسمات السلوكية للحالة. في بعض الحالات AS المرضى لديهم دورات خضعوا لعلاجات مكثفة مماثلة لتعليم موصل التي نفذت في العديد من الأطفال المصابين بالشلل الدماغي. في حين أن بعض الأطفال يخضعون لهذا النوع من العلاج قد أظهرت تحسنا على المدى القصير، على سبيل المثال، في التنقل والاتصالات،
82 لا توجد بيانات حتى الآن تشير إلى أن هذا سوف نقدم الفائدة طويلة الأجل في متلازمة أنجلمان. وهناك أدلة من الآباء والأمهات، وإن كان القولية، ان التدليك والروائح يمكن أن تحسن فرط النشاط والتركيز. اقترحت دراسة بريطانية أن AS تظهر الناس في حاجة إلى التعزيز المستمر لمهاراتهم إذا لم تكن لتفقد لهم. علاج الصرع في AS غالبا ما تكون صعبة، وخاصة في السنوات الأولى،
15، 83 وينبغي التماس المشورة من طبيب أعصاب الأطفال. يمكن أن المضبوطات تتخذ أشكالا عديدة وأخذ مقطع فيديو قصير من المضبوطات المشتبه تظهر للأعصاب مفيد. AS الأطفال لديهم مشاكل التنموية العالمية، ولكن المشكلة أكثر وضوحا هو مع اكتساب اللغة. لا توجد طريقة التواصل واحد يعمل بشكل أفضل في AS ذلك ينبغي بذل كل محاولة لإيجاد نظام الاتصالات الذي يعمل لحساب فرد AS الطفل. لا تزال هناك بعض الأطفال الذين جدا مهارات التواصل مهما المدخلات التي يتلقونها من الآباء والمعالجين محدودة. AS الأطفال لديهم مهارات اجتماعية جيدة نسبيا وتناسب بشكل جيد مع الآخرين ضمن أقرانهم في نفس المجال.يمكن الكامنة الفضول والطفولة فرط النشاط غالبا ما تثير مشاكل الإدارة، واضطراب النوم هي واحدة من أهم القضايا لآباء الأطفال الصغار. العديد من السلوكيات المشكلة المرتبطة حالة يمكن تحسينها من خلال اتباع نهج ثابت، مع مساعدة من المعالج السلوكي إذا لزم الأمر. في البالغين، مع ظهور مؤخرا من مشاكل سلوكية ينبغي النظر في إمكانية ارتداد المريء.
وقد تم تجميع كمية كبيرة من المعرفة على مدى السنوات ال 10 الماضية حول المظاهر السريرية، والتاريخ الطبيعي، والآليات الجينية التي تساهم في متلازمة أنجلمان. كما تم إجراء دراسات للحالة من قبل مجموعة متنوعة من المهنيين العاملين في إدارة المرضى الذين يعانون من AS. العديد من البلدان في جميع أنحاء العالم الآن لديها مجموعات الدعم لذوي AS وأسرهم، وهذه المجموعات قد أنتجت كمية كبيرة من المعلومات عن مختلف جوانب الوضع ويكون في كثير من الحالات كان فعالا في الجمع بين الوالدين والمهنيين العاملين في مجال رعاية AS الناس . منظمة مظلة دولية موجودة الآن
(www.iaso.com) ويمكن أن توجه الآباء والمهنيين لمجموعة متنوعة من AS الموارد والمعلومات حول مواطن الفرد AS المجموعات. ASSERT، فريق الدعم UK، لديها خط هاتف مجاني للآباء والأمهات وخط المساعدة المهنية.
 « آخــر المشـاركــات »
« آخــر المشـاركــات » 10-14-2015, 04:24 PM
10-14-2015, 04:24 PM






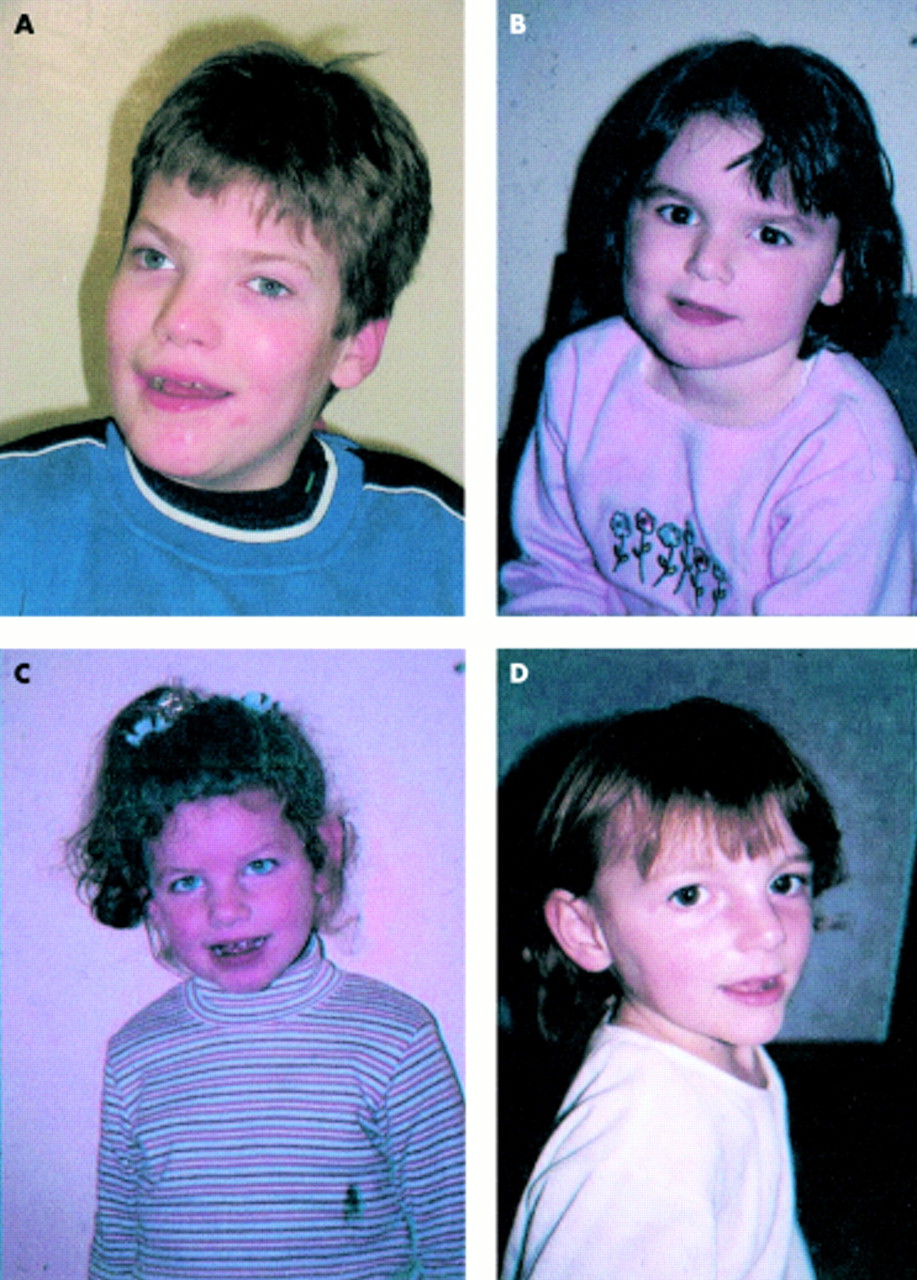




 العرض الشجري
العرض الشجري